
 |
|  ISSN: 2997-321X Doi: 10.47991/2997-321X/AJCRR-109
ISSN: 2997-321X Doi: 10.47991/2997-321X/AJCRR-109
A Rare Treatable Cause for Cognitive Impairment Study Type: Case Report
Author(s): Haider Alrubaiee1*, Seyed Mohammad2, Krystian Figlewsi3, Agnieszka Monika Delekta4, Lorenz Oppel3, Boris Modrau3
1Department of Neurophysiology, Aarhus University Hospital, Denmark.
2Department of Radiology, Holbaek hospital, Denmark.
3Neurology department, Aalborg University hospital, Denmark.
Haider Alrubaiee
Department of Neurophysiology, Aarhus University Hospital, Denmark. Email: h.alrubaiee@rn.dk; Co-author(s): Mtahami@gmail.com (SK); Krfi@rn.dk (KF); agndel@rm.dk (AMD); loop@rn.dk (LO); boris.modrau@rn.dk (BM)
Citation: Alrubaiee H, Mohammad S, Figlewsi K, Delekta AM, Oppel L, Modrau B (2024) A Rare Treatable Cause for Cognitive Impairment Study Type: Case Report. American J Cas Rep Rev: AJCRR-109.
Received: 06 June, 2024
Accepted: 12 June, 2024
Published: 18 June, 2024
Abstract
Background: Cerebral Amyloid Angiopathy related inflammation CAA-ri is a rare clinical complication of Cerebral Amyloid Angiopathy CAA, which is characterized by acute or subacute onset of neurological signs and symptoms such as headaches, seizures, dizziness, other focal signs, rapidly progressive cognitive decline as well as behavioral changes and psychiatric symptoms. Potentially reversible condition with treatment.
Case report: We report a 79-year-old man with subacute progressive cognitive impairment, gait and gaze disturbances of 2 weeks duration. Neurological examination revealed pyramidal and cerebellar signs as well as mild cognitive decline in neuropsychological examination. Magnetic resonance imaging of the brain revealed CAA-ri with watershed infarcts. The patient responded well to steroids and immunosuppressive treatment with marked clinical improvement on further consultation´s visits.
Conclusion: CAA-ri diagnosis is supported by Boston Criteria 2, with characteristic changes in the Magnetic Resonance Imaging of the brain and it is potentially reversible with steroids and immunosuppressive therapy.
Keywords: Cerebral Amyloid Angiopathy, Cerebral Amyloid Angiopathy Related Inflammation, Watershed Infract, Cognitive Impairment, Ischemic Stroke, Alzheimer.
Abbreviations
CAA-ri: cerebral amyloid angiopathy related inflammation
ARIA: Amyloid Related Imaging Abnormalities
Anti-MPO: Anti-Myeloperoxidase Antibody
P-ANCA: Perinuclear anti-neutrophil cytoplasmic antibodies.
P-ANA: Plasma antinuclear antibodies.
Anti-GBM: Anti-Glomerular basement membrane antibody.
Anti-PR3: anti-proteinase 3 antibody.
Introduction
Cerebral amyloid angiopathy (CAA) is a common small vessel disease characterized by the deposition of amyloid β (Aβ) protein mainly in the media and adventitia of small- and medium-sized leptomeningeal and cortical blood vessels. CAA can present with acute lobar intracerebral hemorrhage, chronic progressive cognitive decline as well as transient focal neurological episodes [1]. Cerebral amyloid angiopathy-related inflammation (CAA-ri) is a subtype of reversible encephalopathy seen in a subset of patients with cerebral amyloid angiopathy (CAA) presenting primarily with subacute confusion and behavioral changes [1].
CAA-ri is a rare clinical entity, characterized by rapidly progressive cognitive decline, behavioral changes, headache and seizures, and Magnetic Resonance Imaging (MRI) findings highlighting the inflammatory changes at the level of CAA-affected vessel [2].
MRI shows in CAA-ri symmetric or asymmetric, patchy, or confluent T2 hyperintense lesions in the cortex and subcortical white matter, in addition to the presence of microbleeds on T2- weighted gradient echo sequences (T2* or SWI), the typical finding in CAA [1].
We report a patient with symptoms of CAA-ri where MRI showed Amyloid Related Imaging Abnormalities (ARIA) pattern and ischemic lesions in watershed areas. To our knowledge there has not been reported a similar case of CAA-ri with both of mentioned findings on MRI.
Case Presentation
A 79-year-old man with a history of limited visual acuity due to bilateral glaucoma, a newly diagnosed arterial hypertension and a family history of stroke was admitted to the department of neurology due to subacute progressive confusion in form of distractibility and reduced attention, worsening of visual acuity, and gait instability without falls over the last 14-days. There were no complaints of headache, seizures, or memory difficulties, nor complaints of focal neurological deficits.
On the initial neurological examination, the patient was alert, oriented to person, place and time, and with normal speech. The abnormal findings were bilateral gaze-induced nystagmus, dysmetria at finger nose test most prominent on the right side, hypertonia and hyperreflexia grade 4 in the lower extremities, and bilateral extensive plantar response. The gait was slightly unsteady and heel to toe walk was not possible. The sensory and extrapyramidal system was found to be normal. The blood pressure was 170/90 mmHg, equal in both arms, with no evidence of orthostatic hypotension, temperature 36.7.
The routine blood samples showed no evidence of infectious or metabolic causes, especially normal lever function test and plasma ammonium level. Serum anti-MPO, P-ANCA, P-ANA, P-GBM and P-PR3 were negative.
The MRI of the brain with and without contrast (Figure 1) showed ischemic lesions in the watershed area in the cerebral hemispheres and the cerebellum, subcortical edema in the right temporal cerebral hemisphere, periventricular white matter lesions, and multiple subcortical microbleeds.
The cerebrospinal fluid was without pleocytosis, but elevated protein of 1.13 g/l (normal range 0.15 to 0.6 g/L), elevated albumin 0.6 g/l (normal range 0.15-0.55), and elevated immunoglobulin G 0.184 (g/l) (normal range 0.0015-0.045) indicating inflammation, with no evidence of viral and bacterial infection emphasized by negative PCR for viral DNA/RNA and culture and sensitivity testing. Oligoclonal bands were not present.
Duplex ultrasound examination of both carotid arteries showed mild atherosclerosis without significant stenosis (>50%), as well as normal flow in both vertebral arteries and the 5-days telemetry monitoring without atrial fibrillation.
The neuropsychological evaluation with continuous subtraction test, Boston naming test and word mobilization test showed a performance slightly below expectation. However, other parameters such as orientation, information and verbal memory tests were as expected according to his age. In summary, slight cognitive impairment was concluded based on the age matched performance.
On ophthalmologic examination, the patient’s acute worsening of vision could not be explained by his otherwise well-treated chronic simple glaucoma.
In summary, the diagnostic work up pointed towards a cerebrovascular disease with inflammation.
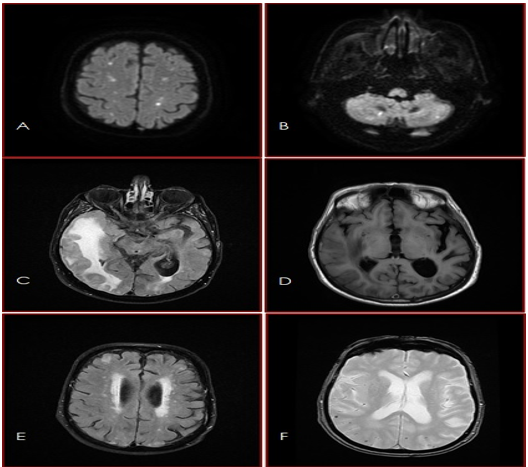
Figure 1: MRI of the brain: The diffusion weighted sequence DWI (A&B) showed ischemic lesions in the watershed area in the cerebral hemispheres and the cerebellum. The T2-FLAIR (C) and the T1-sequence (D) showed subcortical edema in the right temporal cerebral hemisphere. The T2-FLAIR sequence (E) showed periventricular white matter lesions (FAZEKAS 2). The T2*-sequence (F) showed multiple subcortical microbleeds.
After careful diagnostic workup to exclude other causes, and primarily based on the brain imaging, our final diagnosis was cerebral amyloid angiopathy related inflammation (CAA-ri). The patient´s MRI fulfilled the criteria of probable CAA according to Boston criteria 2.0 (citation) and the extensive subcortical edema in combination with the subacute clinical course suits with CAA-ri.
Immunosuppressive therapy was started with 4 days of high dose methylprednisolone (1000 mg intravenous) followed by 96 mg per oral with gradual tapering over the next 8 weeks. Additional antiplatelet therapy with 75mg acetylsalicylic acid was started.
At 8-week control consultation, marked clinical and radiological improvement (figure 2A&B). At that timepoint, medical treatment consisted of Methylprednisolone tablet 8 mg/day as well as acetylsalicylic acid tablet 75 mg/day. A steroid sparing treatment with Azathioprine tablet 100 mg daily then was introduced.
At 6 months control, the patient had no neurological complaints. A new control-MRI was performed (Figure C&D) that showed complete regression of brain edema.
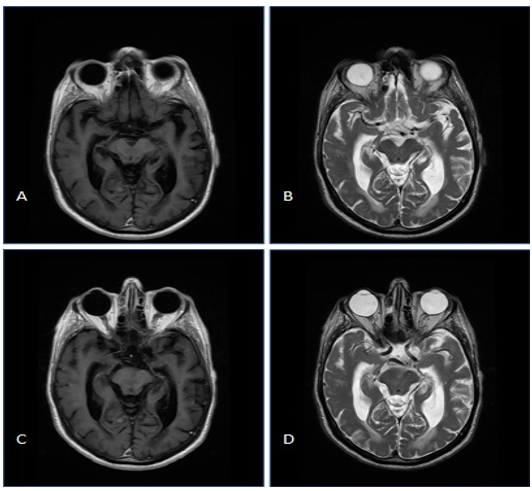
Figure 2: MRI of the brain: Contrast enhanced T1 and T2 sequence at 8 weeks (A&B) and at 6 months (C&D) showed significant regression of edema in the right temporal lobe and full regression of the brain edema respectively.
Discussion
Cerebral Amyloid Angiopathy (CAA) is a disease of small to medium sized cerebral vessels, defined by pathological disposition of amyloid-beta (Aβ) peptides in the media and adventitia of cortical, subcortical, and leptomeningeal arteries, arterioles, and capillaries. The vessels involved get acellular thickening, fibrinoid necrosis, focal wall fragmentation, and morphological dilatation, segmentally with microaneurysm formation [3]. The whole process leads to weakening of the vessels, resulting in recurrent microbleeds, lobar intracerebral hemorrhages and microinfarcts, which eventually clinically lead to cognitive decline and dementia [4].
A rare complication to CAA is a potentially reversible acute / subacute encephalopathy (1), so called Cerebral Amyloid Angiopathy-related inflammation (CAA-ri) [6]. An autoimmune reaction against the Aβ-deposits in the blood vessels wall is the suspected etiology.
Symptoms of CAA-ri are acute or subacute onset of neurological signs such as headaches, confusion, seizures, dizziness, focal signs, rapidly progressive cognitive decline as well as behavioral changes and psychiatric symptoms [7].
MRI findings include cortical/subcortical, symmetric, or asymmetric patchy or confluent lesions on T2 weighted or FLAIR sequences, and signs of CAA in form of microbleeds on T2- weighted gradient echo sequences [5]. These changes were named Amyloid Related Imaging Abnormalities – Edema (ARIA-E) by the Alzheimer’s Association Research Round Table Workgroup [9].
A definite diagnosis of CAA-ri can only be made by biopsy from cerebral and leptomeningeal arteries. This procedure has risks, and a negative brain biopsy does not necessarily rule out CAA-ri [8].
Chung et al, proposed the Boston criteria in 2011 to diagnose CAA-ri. Probable CAA-ri is made when patient 1. Is older than 40 years old 2. Debuted with symptoms in an acute or subacute pattern 3. Have at least one of the symptoms as headache, focal neurological signs, mental status or behavioral change or seizures 4.Have patchy or confluent hyperintensity on T2 or FLAIR, which is usually asymmetric, and could be with or without mass effect and leptomeningeal or parenchymal enhancement 5.Has evidence of a preexisting CAA on MRI in form of multiple cortical and subcortical hemorrhages or microhemorrhages and/or recent or past lobar hemorrhage 6. Has not any neoplastic, infectious, or other causes that can explain the symptoms [10].
These criteria were modified in 2016 by Auriel et al., who among others added the fact that the clinical symptoms in CAA-ri could also have a chronic onset (11). The sensitivity to diagnose CAA by these Boston criteria 2.0 was confirmed in a neuropathology study published I 2022 [11].
The diagnosis of CAA-ri in our patient is based on age (79 years old), subacute onset of symptoms, and an abnormal MRI with CAA-ri-typical findings. The diagnosis is supported by the fact that our patient responded to immunosuppressive therapy, as expected in inflammatory conditions. The increased number of microbleeds in control MRI might be due to the nature of the disease or attributed to steroid therapy [6].
Considering the differential diagnosis, the patient’s CSF with normal WBC´s count and lack of headache contradict primary CNS vasculitis [12] and infection. Rheumatological serological tests exclude most common rheumatological diseases (thereby secondary vasculitis). The negative oligoclonal band and the high albumin with almost normal IgG/albumin log ratio suggests cerebral inflammation comparable with CAA-ri [8] or demyelinating diseases.
The MRI also showed acute and sub-acute ischemic lesions in the watershed area of the cerebrum and cerebellum. This type of ischemic lesions is traditionally believed to be related to hemodynamic impairment. No cardiac arrythmia, significant carotid atherosclerosis or orthostatic hypotension were found on the further work-up in our patient. Therefore, we argue that this pattern of watershed ischemic lesions is related to CAA. This pattern of infarction is also described in patients with Alzheimer’s disease [12]. Common with CCA and Alzheimer’s disease is the congophilic angiopathy. In CAA, Aβ 40 and 42 amyloid peptides are predominantly found in the vessel wall, but also found in senile plaques in Alzheimer’s disease (1). Thus, the pathogenesis of cortical watershed microinfarcts in Alzheimer’s disease and CAA might be similar. Of interest in our case, no further acute infarction at follow-up was seen after the successful immunosuppressive therapy of the inflammation component of CAA-ri.
Result
The study demonstrates a rare case of cerebral amyloid angiopathy related inflammation with good response to steroids and steroid sparing immunosuppressants.
Conclusion
Our patient did fulfill the Boston 2.0 criteria for probable CAA, the presentation was typical for CAA-ri, and responded well to immunosuppressive therapy. As CAA-ri is a rare disease, our case might contribute to the growing understanding of this condition and might contribute to recalling the clinical and imaging features.
Conflict of Interest
The authors declare that there is no conflict of interest.
Acknowledgement
All authors contributed equally to this manuscript. All authors approved the final draft.
Source of funding: The authors received no specific grant from any funding agency in public commercial, or not-for-profit sectors.
References
Kirshner HS, Bradshaw M. The Inflammatory Form of Cerebral Amyloid Angiopathy or “Cerebral Amyloid Angiopathy-Related Inflammation” (CAARI). Curr Neurol Neurosci Rep. 2015 Aug;15(8):54.
Du, Y., Liu, C., Ma, C. et al. Cerebral amyloid angiopathy-related inflammation: a case report presenting with a rare variant in SORL1 gene. BMC Neurol 19, 97 (2019).
Vinters HV. Cerebral amyloid angiopathy a critical review. 1987; 18:311–324.
Karol Adam Makarewicz, Karolina Zaryczańska, Karolina Machowska-Sempruch, Anna Bajer-Czajkowska, Piotr Gołofit, Ewa Gabrysz-Trybek, Przemysław Nowacki. Cerebral amyloid angiopathy-related inflammation (CAARI): case report. Folia Neuropathol 2019; 57 (2): 205-210
Weber SA, Patel RK, Lutsep HL. Cerebral amyloid angiopathy: diagnosis and potential therapies. Expert Rev Neurother. 2018 Jun;18(6):503-513.
Wu JJ, Yao M, Ni J. Cerebral amyloid angiopathy-related inflammation: status and future implications. Chin Med J (Engl). 2021;134(6):646-654. Published 2021 Feb 23.
Sperling RA, Jack CR, Black SE, et al. Amyloid-related abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer’s Association Research Round Table Workgroup. Alzheimers Dement 2011; 7:367–85.
Chung KK, Anderson NE, Hutchinson D, et al. Cerebral amyloid angiopathy related inflammation: three case reports and a review. J Neurol Neurosurg Psychiatry. 2011; 82:20–26.
Auriel E, Charidimou A, Gurol ME, et al. Validation of Clinicoradiological Criteria for the Diagnosis of Cerebral Amyloid Angiopathy–Related Inflammation. JAMA Neurol. 2016;73(2):197–202. doi:10.1001/jamaneurol.2015.4078
Beuker C, Schmidt A, Strunk D, et al. Primary angiitis of the central nervous system: diagnosis and treatment. Ther Adv Neurol Disord. 2018;11: 1756286418785071. Published 2018 Jul 9. doi:10.1177/1756286418785071.
The Boston criteria version 2.0 for cerebral amyloid angiopathy: a multicentre, retrospective, MRI–neuropathology diagnostic accuracy study, The Lancet Neurology, Volume 21, Issue 8, August 2022, Pages 714-725
Cerebral Hypoperfusion Generates Cortical Watershed Microinfarcts in Alzheimer Disease Oda-Christina Suter, Thanomphone Sunthorn, Rudolf Kraftsik, Joel Straubel, Pushpa Darekar, Kamel Khalili and Judith Miklossy, Originally published1 Aug 2002. 2002; 33:1986–1992